1、简介
Shannon, Simpson, and Fisher diversity indices and species richness.
2、使用语法
diversity(x, index = "shannon", groups, equalize.groups = FALSE,
MARGIN = 1, base = exp(1))
simpson.unb(x, inverse = FALSE)
fisher.alpha(x, MARGIN = 1, ...)
specnumber(x, groups, MARGIN = 1)3、参数解释
3.1 diversity()
- 作用: 计算样本的生物多样性指数。
- 参数:
- x: 包含生物数据的矩阵或数据框。
- index: 指定要计算的生物多样性指数,例如 Shannon、Simpson 、Invsimpson等。
- groups: (可选)分组变量,用于根据其值计算指数。
- equalize.groups: (可选)逻辑值,指示是否在计算指数之前均衡组的样本数。
- MARGIN: 指示应用函数的维度,1 表示按行应用,2 表示按列应用。
- base: (可选)用于 Shannon 指数计算的底数。
- 返回值: 返回一个包含生物多样性指数的向量或矩阵。
3.2 simpson.unb()
- 作用: 计算样本的 Simpson 无偏估计。
- 参数:
- x: 包含生物数据的矩阵或数据框。
- inverse: (可选)逻辑值,指示是否返回 Simpson 的逆指数(即倒数)。
- 返回值: 返回 Simpson 未偏估计的值或其逆。
3.3 fisher.alpha()
- 作用: 计算样本的 Fisher's alpha 指数。
- 参数:
- x: 包含生物数据的矩阵或数据框。
- MARGIN: 指示应用函数的维度,1 表示按行应用,2 表示按列应用。
- ...: 其他参数,用于传递给底层函数。
- 返回值: 返回 Fisher's alpha 指数的值。
3.4 specnumber()
- 作用: 计算样本的物种数或群落中的物种丰富度。
- 参数:
- x: 包含生物数据的矩阵或数据框。
- groups: (可选)分组变量,用于根据其值计算物种数。
- MARGIN: 指示应用函数的维度,1 表示按行应用,2 表示按列应用。
- 返回值: 返回样本中的物种数。
4、举例
代码来自于:[Package vegan version 2.6-4 Index],在Rstudio中可找到这个代码。
这里对生态数据集BCI进行了一系列生物多样性分析,并进行了一些统计计算和绘图,以了解不同栖息地之间的物种多样性和差异。
# 加载BCI和BCI.env数据集
data(BCI, BCI.env)
# 计算整体生物多样性(Shannon指数)
H <- diversity(BCI)
# 计算Simpson指数
simp <- diversity(BCI, "simpson")
# 计算Simpson逆指数
invsimp <- diversity(BCI, "inv")
# 计算无偏估计的Simpson指数
unbias.simp <- simpson.unb(BCI)
# 计算Fisher's alpha指数
alpha <- fisher.alpha(BCI)
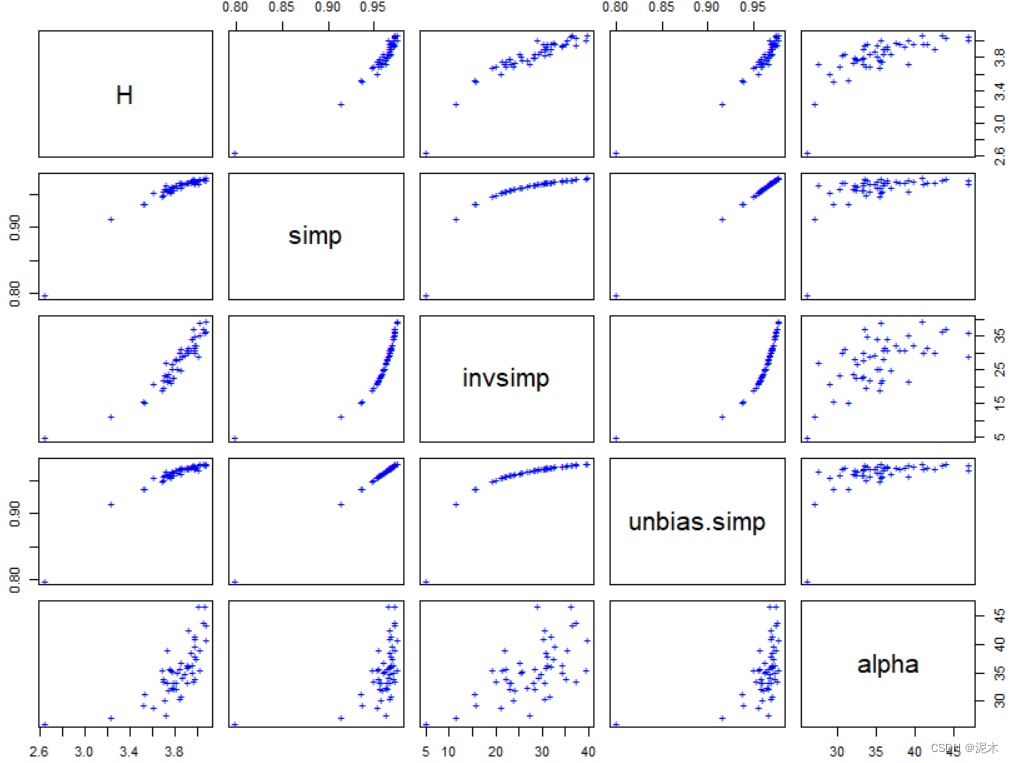
# 绘制散点图矩阵以比较各指数
pairs(cbind(H, simp, invsimp, unbias.simp, alpha), pch="+", col="blue")
# 物种丰富度(S)和Pielou's均匀度(J)的计算:
S <- specnumber(BCI) # 或者使用 rowSums(BCI > 0)
J <- H/log(S)
# beta多样性定义为gamma/alpha - 1:
# alpha是组内平均物种数,gamma是组内总物种数
alpha <- with(BCI.env, tapply(specnumber(BCI), Habitat, mean))
gamma <- with(BCI.env, specnumber(BCI, Habitat))
gamma/alpha - 1
# 类似的计算可以使用Shannon多样性指数
alpha <- with(BCI.env, tapply(diversity(BCI), Habitat, mean)) # 平均值
gamma <- with(BCI.env, diversity(BCI, groups=Habitat)) # 合并值
# 基于Shannon指数的加性beta多样性
gamma-alpha
Run example:
参考:Package vegan version 2.6-4