目录
文献阅读:基于遗传算法的PM2.5时间序列预测深度学习模型超参数优化
摘要
本周阅读的论文《Genetic algorithm‑based hyperparameter optimization of deep learning models for PM2.5 time‑series prediction》介绍了一种创新方法,通过遗传算法(GA)优化深度学习模型的超参数,以提高对空气中PM2.5浓度的预测精度。针对学习率、丢弃率、隐藏层数量及每层单元数、激活函数、损失函数和优化器等关键超参数进行优化,以期达到模型训练的最佳性能。鉴于传统统计和机器学习模型在处理非线性关系和序列数据方面的局限性,研究采用LSTM、RNN和GRU等深度学习技术。此外,论文还讨论了数据预处理的重要性,包括识别并处理缺失、错误或噪声数据,如通过插值方法填补缺失值,利用极值检测规则记录极端数据点,并采用最小-最大缩放法标准化数据,确保模型训练的稳定性和准确性。
Abstract
The paper I read this week, "Genetic algorithm based hyperparameter optimization of deep learning models for PM2.5 time-series prediction," introduces an innovative method that optimizes the hyperparameters of deep learning models through genetic algorithm (GA) to improve the accuracy of predicting PM2.5 concentration in the air. Optimize key hyperparameters such as learning rate, dropout rate, number of hidden layers and units per layer, activation function, loss function, and optimizer to achieve optimal model training performance. Due to the limitations of traditional statistical and machine learning models in handling nonlinear relationships and sequence data, deep learning techniques such as LSTM, RNN, and GRU were adopted in the study. In addition, the paper also discusses the importance of data preprocessing, including identifying and processing missing, erroneous, or noisy data, such as filling in missing values through interpolation methods, recording extreme data points using extreme value detection rules, and standardizing data using the minimum maximum scaling method to ensure the stability and accuracy of model training.
文献阅读:基于遗传算法的PM2.5时间序列预测深度学习模型超参数优化
一、现有问题
- PM2.5预测存在数据具有非线性关系,需要处理大数据集,以及研究人员必须处理噪声、缺失或不完整的数据的难题。传统统计学与机器学习方法在空气质量预报中虽有广泛应用,但它们在捕捉非线性关系和处理序列数据方面存在局限性。特别是在大都市环境中,由于高人口密度和发达工业,空气质量监测尤为重要。然而,传感器错误可能导致采集的空气质量指标(如PM2.5、PM10等)数据不完整或有误,这进一步增加了预测难度。
- 在机器学习模型中,有两种类型的参数。一个是反映数据特征的模型参数。第二种是超参数,它影响学习质量和算法性能,在训练过程中不会发生变化。因此需要仔细研究的不同技术来配置超参数以达到更好的性能。
二、提出方法
本研究提出了一种基于遗传算法的超参数优化(HPO)方法,利用基于遗传算法的方法来优化LSTM、GRU和RNN深度学习神经模型的超参数。该研究的目的是在一个定义的搜索空间中找到一组超参数的最佳组合,而无需研究人员的经验要求,提升深度学习模型在PM2.5浓度预测中的性能和预测精度。
研究贡献
- 提出了一种适用的基于遗传算法的HPO技术,用于深度神经网络模型,以实现对PM2.5数据的高预测性能。
- 所选数据是取自伊斯坦布尔大都市的实际数据集,由许多不完整、不正确或缺失的数据和离群值组成,采用数据挖掘技术为深度学习模型准备数据的预处理。
- 该数据集包括空气污染物以及气象数据,组合数据集通过24小时历史数据的时间窗口进行训练,以预测下一小时的PM2.5浓度。
三、方法论
1、HPO(猎人猎物算法)
算法思想
HPO模拟了自然界中动物的捕猎过程,该算法的基本假设是:在猎人寻找猎物的场景中,猎人将大概率的选择一个远离群体的猎物(远离平均群体位置)。猎人找到猎物后就会追逐猎物。与此同时,猎物寻找食物,并在捕食者的攻击中逃脱,到达一个安全的地方,这两个过程中即伴随着猎人位置与猎物位置的更新。根据适应度函数,我们最终认为这个安全的地方(目标搜索位置)是最佳猎物所在的地方,从而完成了整个搜索的过程,下面分别是猎人追击猎物与猎物逃跑过程的示意图。深度学习模型包含大量影响其表现的超参数,例如学习率、丢弃率、隐藏层数量及每层单元数、激活函数、损失函数和优化器等。手动调优这些参数费时且效率低下,而基于遗传算法的HPO能够通过模拟自然选择和遗传机制,在预定义的搜索空间内自动找到最佳超参数组合。这种方法利用种群初始化、选择、交叉变异和适者生存等步骤,不断迭代以优化模型性能。

超参数优化(HPO)算法一般都包括三类:
- 暴力搜索类: 随机搜索(random search)、网格搜索(grid search)、自定义(custom)
- 启发式搜索: 进化算法(Naïve Evolution, NE)、模拟退火算法(Simulate Anneal, SA)、遗传算法(Genetic Algorithm, GA)、粒子群算法(Particle Swarm Optimization, PSO)等自然计算算法、Hyperband
- 贝叶斯优化: BOHB、TPE、SMAC、Metis、GP
算法过程

详细的猎人猎物优化算法可以参考下面这篇博客:
基于猎人猎物优化算法的函数寻优算法_心️升明月的博客-CSDN博客
2、深度学习算法
本研究中应用的深度学习算法包括长短期记忆网络(LSTM)、门控循环单元(GRU)和循环神经网络(RNN)。这些模型因其在处理序列数据上的强大能力,在时间序列预测领域表现出色,能有效捕捉时间序列中的长期依赖关系。在这项研究中,多变量时间序列预测将进行每小时10分钟的PM2.5数据集。PM2.5是目标变量,受当前和先前状态的影响。出于这个原因,RNN及其变体,LSTM和GRU深度学习模型是首选。
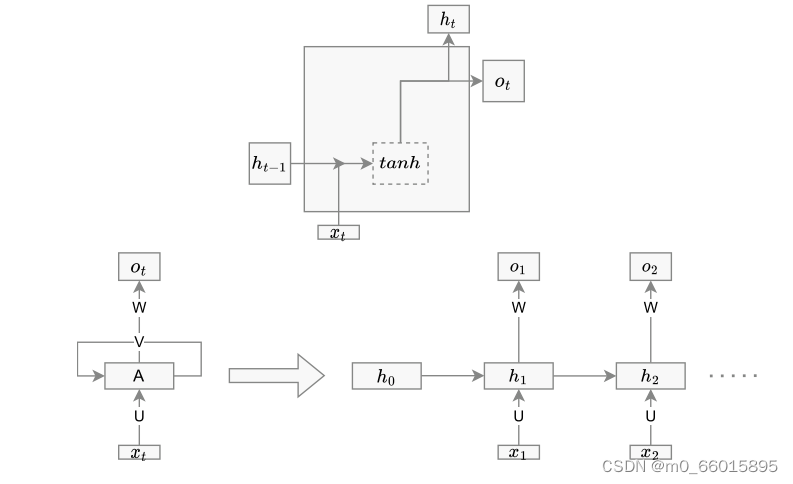
递归神经网络(RNN)
是一种深度神经网络,也是人工神经网络的一种变体,它模仿人脑神经系统,可以在网络中存储以前的信息。与ANN不同,RNN有输入层、隐藏层和输出层。由于观测序列对时间序列分析结果有很大影响,因此RNN可以在训练时有效地发现数据模式。RNN单元结构如下图,节点是单个时间戳上的神经网络单元。X是特征集合,XT是时间t时X的值。Ot是输出,Ht是隐藏状态。输入层连接到隐藏层,隐藏层连接到输出层。这些连接由权重矩阵U、W和V表示:
LSTM
LSTM是RNN的一种变体,为解决序列数据建模中的消失梯度问题而专门设计的,可以克服对神经网络系统的长期依赖,适用于时间序列预测应用。经典的LSTM由三种类型的门组成:输入门、遗忘门和输出门。输入门控制来自存储单元的信息;类似地,输出门控制输出流。
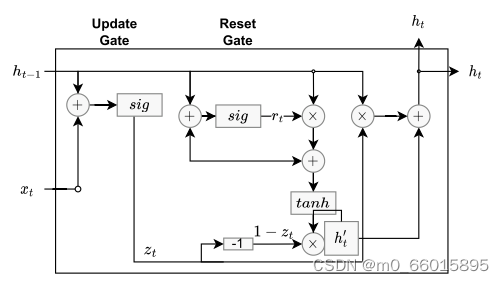
GRU
GRU单元被设计为LSTM的简单变体。GRU只有更新(z)(类似于LSTM中的输入和遗忘门)和重置(r)门。要记住的信息保存在更新门中,其较低的复杂性意味着GRU比LSTM使用更少的参数,并且在训练期间需要更少的计算能力。
3、GA(遗传算法)
遗传算法是一种启发式搜索策略,它模仿生物进化过程中的自然选择和遗传原理。在超参数优化场景中,遗传算法通过创建一个初始解集(即种群),并基于每个解(即一组超参数配置)的适应度(模型在验证集上的表现)进行评估。之后,通过选择、交叉和变异操作生成新的解,逐步迭代,直到达到预定的停止条件(如最大迭代次数或性能阈值)。染色体(个体)表示遗传算法中随机生成的解决方案,并具有适应度值。种群由一个以上的染色体组成,通过选择、交叉和变异算子,将根据种群中的适应值排序的染色体传递到下一个种群,具有高适应值的染色体将高信息传递给下一个种群。因此,优良染色体的遗传特性得以保留,发育得以持续。染色体表示和适应度函数的确定对算法的性能有很大影响。

在这项研究中,深度学习算法的预测性能被确定为染色体的适应度值。在研究步骤中,首先获得由气象和空气质量数据组成的数据集。然后,进行数据预处理步骤。分析利用遗传算法产生的染色体超参数结构进行训练的性能,在将性能分配为染色体的适应度之后,应用遗传算法的过程。所提出的GA优化深度学习算法超参数的总体框架如图所示。
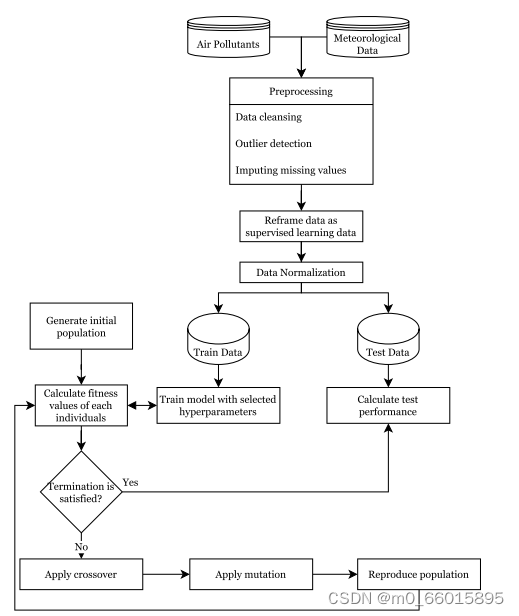
应用基本的四个步骤来采用合适的GA设计来预测PM2.5浓度,GA算法的过程如下:
- 初始化:在GA中首先生成随机初始种群,种群大小取决于算法类型。在数据准备阶段之后,为初始群体随机生成10条染色体,使用测试集的MSE性能计算每个染色体的适应度。此外,在训练数据集上测试模型,并比较测试和训练性能以计算适应度。在根据染色体的适应度值对染色体进行排序之后,生成了种群的最佳、平均和最差染色体,这些染色体将在以下步骤中使用。
- 选择和繁殖:选择运算符选择适应度较高的染色体来繁殖下一代的新染色体,根据轮盘赌选择技术,高适应度染色体比低适应度染色体有更多被选择的机会。除了轮盘赌之外,每一代都会创建一个随机个体,以获得更大的遗传多样性。此外,对于精英选择,最好的个体会传递给下一代,以避免失去上一个群体中表现最好的组合,并确保遗传算法始终处于比上一个群体更好的最低点。由于在算法过程中有可能淘汰最佳个体,因此精英主义可以保留最佳个体。最后,假设群体中有多个相同的染色体(具有相同的超参数)。在这种情况下,这些个体应该被淘汰,为种群生成一个新的随机染色体。在这项研究中产生了50个世代,使用上述选择和繁殖方法。
- 交叉:交叉是在选择后以一定的速率进行的,称为交叉率。一个单一的切割点允许交配池的染色体从一个特定的点杂交。基因变化发生在两个被选择的个体之间,以确保种群的多样性。在本研究中,对于交叉群体,根据以下可能性选择六条染色体:[0.3,0.2,0.15,0.12,0.10,0.07,0.03,0.02,0.006,0.004]。每个个体在每个世代中用于交换的选择可能性保持恒定。因为在轮盘赌选择中,染色体之间的适应度差异将朝着最后一代减少。
- 突变:在突变步骤中,一个染色体基因被简单地替换为可能的值,它根据预定的突变率执行,提出了一种避免陷入局部最优解的变异算子,变异群体的形成采用多点变异技术。在这项研究中,根据前面提到的突变群体的可能性选择染色体,变异算子中需要考虑和控制的基因是隐藏层大小和深度学习算法。如果突变基因是其中之一,则进行基因控制以保护染色体的可行性。
轮盘赌选择(Roulette Wheel Selection):是一种回放式随机采样方法。每个个体进入下一代的概率等于它的适应度值与整个种群中个体适应度值和的比例。
本研究采用单点交叉与多点变异。下图表示每个基因的染色体结构和搜索空间,以及交叉和变异操作符。


五、研究实验
1、数据集
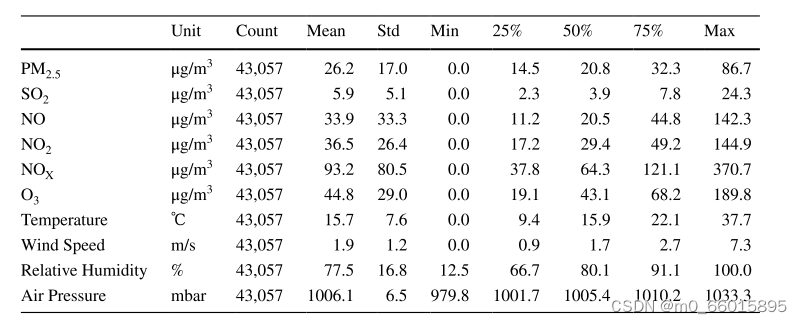
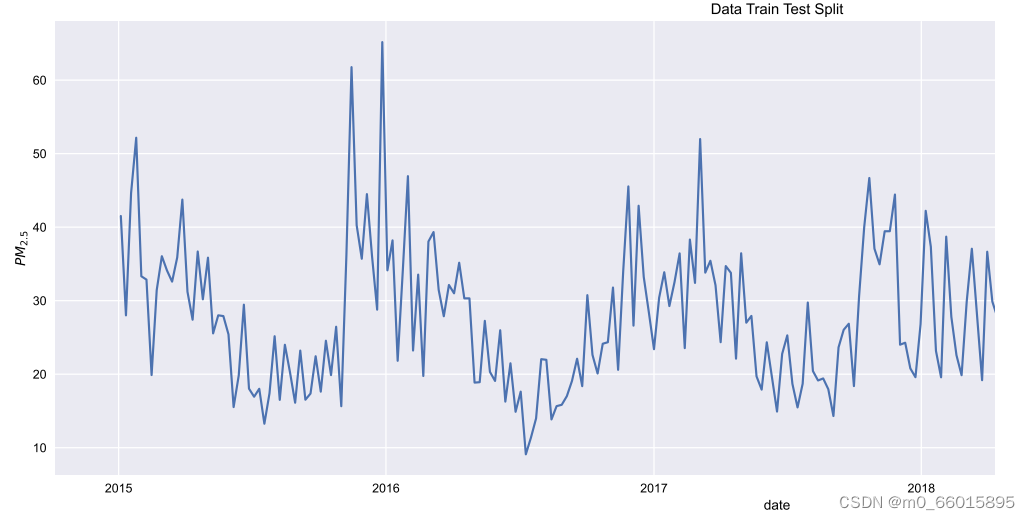
实验研究的数据集是在2015年1月1日至2019年11月30日期间从伊斯坦布尔观测中心收集的,其中气象数据也包括在数据集中。原始数据集由43058个样本组成,包括PM 2.5,SO2,NO,NO2,NO2,NOx,NO3,温度,风速,相对湿度和气压特征。数据集的前70%用作训练集,30%用作测试集。
下图为2015-2019年每周观测到的PM2.5浓度的时间序列
2、评估指标
评估指标包括平均绝对误差(MAE)、平均绝对百分比误差(MAPE)、均方误差(MSE)、决定系数(R²)和均方根误差(RMSE)。这些指标不仅衡量了预测值与实际值之间的偏差程度,还反映了模型的整体拟合质量。特别是,R²值接近1表示模型拟合度好,而低的MAE和RMSE则意味着预测误差小。

3、实验过程
实验首先定义了深度学习模型的超参数空间,包括学习率、丢弃率、隐藏层单元数、激活函数、损失函数和优化器等。然后,通过遗传算法进行超参数优化,这是一种基于自然选择和遗传机制的全局优化策略。算法从初始随机生成的超参数组合出发,通过评估模型在验证集上的性能,执行选择、交叉和变异操作,逐渐进化出更优的超参数组合。实验中,对比了优化前后的模型性能,以及与随机搜索和默认设置下的模型表现。
4、实验结果
遗传算法在超参数优化上的有效性
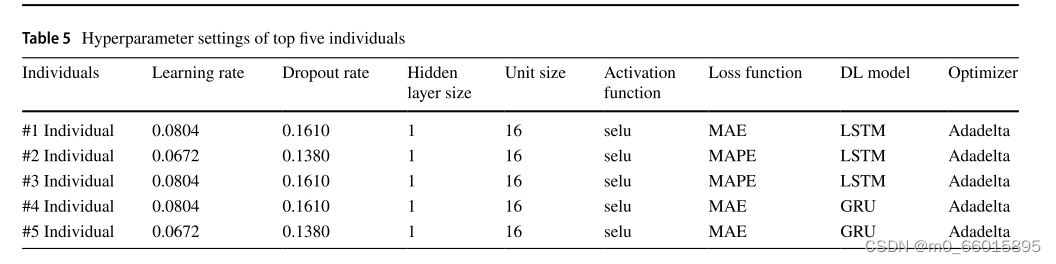
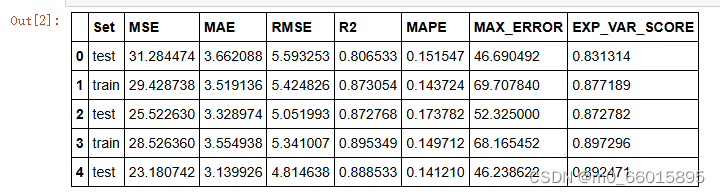
表5和表6给出了遗传算法产生的种群中前五个个体的比较结果。最佳个体的隐藏层数为1,激活函数为“selu”,优化器为Adadelta。遗传算法产生的最佳个体的适应度值为20.5611。优化后的模型在测试集和训练集上均取得了较高的EVS(解释方差得分),较低的MAE、ME、MSE和RMSE,以及接近于1的R²值,证明了遗传算法在超参数优化上的有效性。特别地,个体#3以最低的MAPE和ME在测试集中表现最好,突显了selu激活函数和Adadelta优化器搭配的优越性。


比较遗传算法的性能
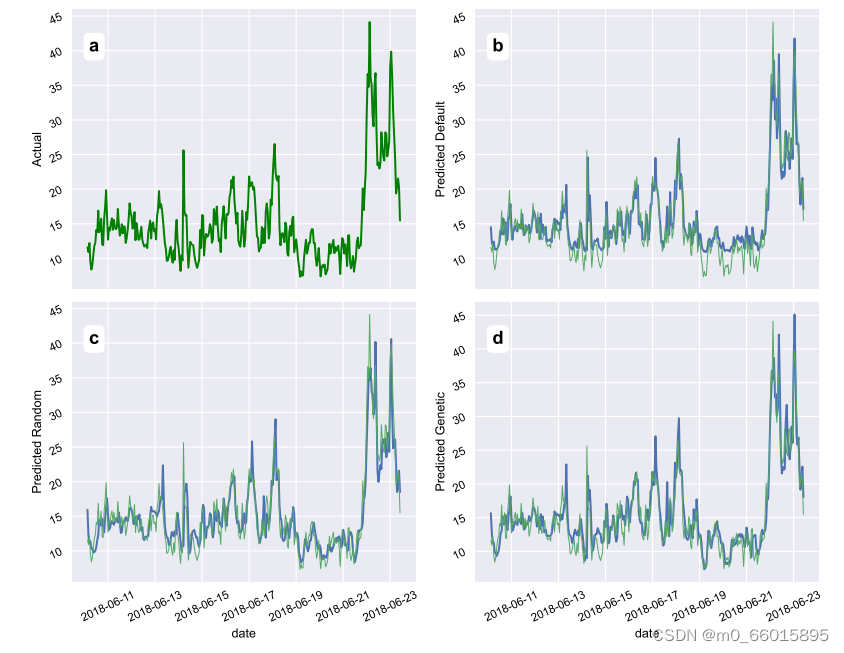
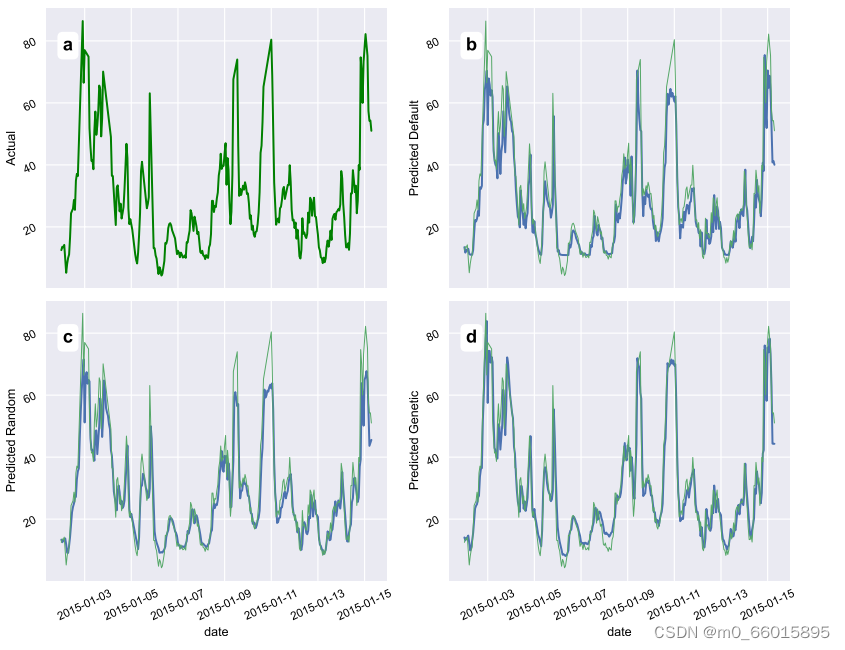
随机搜索算法通过在超参数之间进行随机试验来获得结果,默认超参数表示Keras库为训练提供的默认超参数值,当使用最佳个体的超参数训练PM2.5时间序列预测模型时,使用预测值的测试和训练如图所示,其中(B)默认参数设置、(c)随机搜索设置、(d)基于遗传算法的设置。预测结果表明,遗传算法具有上级的优越性。
六、代码实现
遗传算法
import codecs
import time
import numpy as np
import pandas as pd
import papermill as pm
import pickle
# hiperparameter list
hyper_parameters = {'epoch': 60, 'batch_size': 120, 'layer_size': [1, 2, 4], 'unit_size': [8, 16, 32],
'activations': ['relu', 'sigmoid', 'softmax', 'softplus', 'softsign', 'tanh', 'selu', 'elu'],
'loss_functions': ['mse', 'mape', 'mae'], 'models': ['GRU', 'LSTM', 'SimpleRNN'],
'optimizers': ["SGD", "RMSprop", "Adam", "Adadelta", "Adagrad", "Adamax", "Nadam", "Ftrl"]}
learning_rate_ranges = [0.0001, 0.09]
dropout_rate_ranges = [0.1, 0.5]
ga_parameters = {'max_num_iteration': 50,
'population_size': 10,
'crossover_chromosome_size': 6,
'mutation_chromosome_size': 3,
'crossover_rate': 0.6,
'mutation_rate': 0.3, 'crossover_point_size': 1, 'mutation_point_size': 3,
'prob_chr': [0.3, 0.2, 0.15, 0.12, 0.10, 0.07, 0.03, 0.02, 0.006, 0.004]}
class Chromosome:
def __init__(self, chr_id=0):
"""
:param chr_id: Kromozomun çağrılması için değer
"""
self.chr_id = chr_id
self.generation_number = 0
self.genes = []
self.test_MSE = 0
self.train_MSE = 0
self.test_MAE = 0
self.train_MAE = 0
self.test_RMSE = 0
self.train_RMSE = 0
self.test_R2 = 0
self.train_R2 = 0
self.test_MAPE = 0
self.train_MAPE = 0
self.test_MAX_ERROR = 0
self.train_MAX_ERROR = 0
self.test_EXP_VAR_SCORE = 0
self.train_EXP_VAR_SCORE = 0
self.fitness = 0
def __str__(self):
return self.genes.__str__()
def __repr__(self):
return self.genes.__str__()
def __eq__(self):
return self.genes.__str__()
def generate_genes(self, SEED):
np.random.seed(SEED + int(self.chr_id)) # sabitleme için gerekebilir.
self.genes = []
self.genes.append(np.around(
np.random.uniform(learning_rate_ranges[0], learning_rate_ranges[1]),
6)) # learning rate
self.genes.append(np.around(
np.random.uniform(dropout_rate_ranges[0], dropout_rate_ranges[1]),
3)) # dropout rate
self.genes.append(np.random.choice(hyper_parameters['layer_size'])) # layer size
self.genes.append(np.random.choice(hyper_parameters['unit_size'])) # unit size
self.genes.append(np.random.choice(hyper_parameters['activations'])) # activation function
self.genes.append(np.random.choice(hyper_parameters['loss_functions'])) # loss function
self.genes.append(list())
if self.genes[2] == 1:
self.genes[6].append(np.random.choice(hyper_parameters['models']))
elif self.genes[2] == 2:
self.genes[6].append(np.random.choice(hyper_parameters['models'], size=2).tolist())
elif self.genes[2] == 4:
self.genes[6].append(np.random.choice(hyper_parameters['models']))
self.genes.append(np.random.choice(hyper_parameters['optimizers'])) # optimizer
def change_genes(self, liste):
"""verilen l listesini chromosome genleri olarak alır"""
self.genes = []
self.genes = liste.copy()
def calculate_fitness(self, notebook, epoch, batch_size):
print("Çalıştırılan genler", self.genes)
model_number = str(self.generation_number) + '_' + str(self.chr_id)
pm.execute_notebook('{}.ipynb'.format(notebook),
'notebooks/{}_{}.ipynb'.format(self.generation_number, self.chr_id),
parameters=dict(model_number=model_number, learning_rate=np.float64(self.genes[0]),
dropout_rate=np.float64(self.genes[1]),
layer_size=int(self.genes[2]),
units=int(self.genes[3]),
activation_function=self.genes[4],
loss_function=self.genes[5], epoch=epoch, batch_size=batch_size,
models=self.genes[6], optimizers=self.genes[7]))
df_results = pd.read_csv(
"performances/df_results{}.csv".format(model_number))
self.test_MSE = df_results['MSE'][0]
self.train_MSE = df_results['MSE'][1]
self.test_MAE = df_results['MAE'][0]
self.train_MAE = df_results['MAE'][1]
self.test_RMSE = df_results['RMSE'][0]
self.train_RMSE = df_results['RMSE'][1]
self.test_R2 = df_results['R2'][0]
self.train_R2 = df_results['R2'][1]
self.test_MAPE = df_results['MAPE'][0]
self.train_MAPE = df_results['MAPE'][1]
self.test_MAX_ERROR = df_results['MAX_ERROR'][0]
self.train_MAX_ERROR = df_results['MAX_ERROR'][1]
self.test_EXP_VAR_SCORE = df_results['EXP_VAR_SCORE'][0]
self.train_EXP_VAR_SCORE = df_results['EXP_VAR_SCORE'][1]
self.fitness = min(self.test_MSE, 500)
class Population:
def __init__(self, size, generation):
self.generation = generation
self.size = size
self.chromosomes = [] # 染色体
self.best, self.worst, self.avg = 0, 0, 0
def init_pop(self):
print("initializing population")
for i in range(self.size):
chrom = Chromosome(chr_id=i)
chrom.generation_number = self.generation
self.chromosomes.append(chrom)
def sort_pop(self):
return self.chromosomes.sort(key=lambda x: x.fitness, reverse=False)
def calculate_best_avg_worst(self):
self.sort_pop()
self.best = round(self.chromosomes[0].fitness, 3)
_total = 0
for i, ind in enumerate(self.chromosomes):
_total += ind.fitness
self.avg = max(round(_total / self.size, 2), 500)
self.worst = max(round(self.chromosomes[-1].fitness, 2), 500)
def print_pop(self):
ga_text_file = codecs.open("results\\ga-" + ".txt", "a+")
ga_text_file.write(
"--------------------------------------------------\n")
ga_text_file.write(
"Population #\t{}\tBest\t{}\tAvg\t{}\tWorst\t{}".format(self.generation, self.best, self.avg, self.worst))
ga_text_file.write(
"\n--------------------------------------------------\n")
for i, x in enumerate(self.chromosomes):
ga_text_file.write(
"Individual #\t{}\t{}\tFitness\t{}\ttest_MSE\t{}\ttrain_MSE\t{}\ttest_MAE\t{}\ttrain_MAE\t{}\ttest_RMSE\t{}\ttrain_RMSE\t{}\ttest_R2\t{}\ttrain_R2\t{}\ttest_MAPE\t{}\ttrain_MAPE\t{}\ttest_MAX_ERROR\t{}\ttrain_MAX_ERROR\t{}\ttest_EXP_VAR_SCORE\t{}\ttrain_EXP_VAR_SCORE\t{}\n".format(
i, x.genes, round(x.fitness, 3), round(x.test_MSE, 3), round(x.train_MSE, 3), round(x.test_MAE, 3),
round(x.train_MAE, 3), round(x.test_RMSE, 3), round(x.train_RMSE, 3), round(x.test_R2, 3),
round(x.train_R2, 3), round(x.test_MAPE, 3), round(x.train_MAPE, 3), round(x.test_MAX_ERROR, 3),
round(x.train_MAX_ERROR, 3), round(x.test_EXP_VAR_SCORE, 3), round(x.train_EXP_VAR_SCORE, 3)))
ga_text_file.close()
def reap_pop(self):
self.chromosomes = self.chromosomes[:self.size]
def __str__(self):
return self.chromosomes.__str__()
def __repr__(self):
return self.chromosomes.__str__()
def crossover(p1, p2, points):
if points <= 2: # çaprazlamada 6. genin değişmemesi için önlem aldık.
p1, p2 = list(p1), list(p2)
c1, c2 = p1[:points] + p2[points:], p2[:points] + p1[points:]
else:
p1, p2 = list(p1), list(p2)
c1, c2 = p1[:points] + p2[points:], p2[:points] + p1[points:]
c1[6] = p1[6]
c2[6] = p2[6]
return c1, c2
def sum_to_x(n, x):
values = [0.0, x] + list(np.random.uniform(low=0.0, high=x, size=n - 1))
values.sort()
return [values[i + 1] - values[i] for i in range(n)]
SEED = 1234
nb_name = 'calistirma_genetic'
pop_list = [] # Popülasyon bireyleri
#初始化种群
np.random.seed(SEED)
first_pop = Population(size=ga_parameters['population_size'], generation=0)
first_pop.init_pop()
for ind in first_pop.chromosomes:
ind.generate_genes(SEED=SEED)
ind.calculate_fitness(notebook=nb_name, epoch=hyper_parameters['epoch'],
batch_size=hyper_parameters['batch_size'])
first_pop.sort_pop()
# First Fitness Evaluation
first_pop.calculate_best_avg_worst()
first_pop.print_pop()
pop_list.append(first_pop)
# 初始个体数
np.random.seed(SEED)
first_pop = Population(size=ga_parameters['population_size'], generation=0)
first_pop.init_pop()
for ind in first_pop.chromosomes:
ind.generate_genes(SEED=SEED)
ind.calculate_fitness(notebook=nb_name, epoch=hyper_parameters['epoch'],
batch_size=hyper_parameters['batch_size'])
first_pop.sort_pop()
# 首次适应度评估
first_pop.calculate_best_avg_worst()
first_pop.print_pop()
pop_list.append(first_pop)
import csv
with open('pop_list.csv', 'w', newline='') as f:
writer = csv.writer(f)
writer.writerow(
['generation', 'Individual', 'chromosome_ID', 'genes', 'fitness', 'test_EXP_VAR_SCORE', 'test_MAE', 'test_MAPE',
'test_MAX_ERROR', 'test_MSE', 'test_R2', 'test_RMSE', 'train_EXP_VAR_SCORE', 'train_MAE', 'train_MAPE',
'train_MAX_ERROR', 'train_MSE', 'train_R2', 'train_RMSE'])
for gen_num, pop in enumerate(pop_list):
for chr_rank, chr in enumerate(pop.chromosomes):
writer.writerow(
[gen_num, chr_rank, chr.chr_id, chr.genes, chr.fitness, chr.test_EXP_VAR_SCORE, chr.test_MAE,
chr.test_MAPE, chr.test_MAX_ERROR, chr.test_MSE, chr.test_R2, chr.test_RMSE, chr.train_EXP_VAR_SCORE,
chr.train_MAE, chr.train_MAPE, chr.train_MAX_ERROR, chr.train_MSE, chr.train_R2, chr.train_RMSE])

模型训练
import os
import numpy as np
import random
import matplotlib.pyplot as plt
import pandas as pd
from sklearn.model_selection import train_test_split
from sklearn.metrics import mean_absolute_error, mean_squared_error, mean_absolute_percentage_error, r2_score
from sklearn.metrics import max_error, explained_variance_score
from sklearn.preprocessing import MinMaxScaler
import tensorflow as tf
from datetime import datetime
from tensorflow.keras.models import Sequential
from tensorflow.keras.layers import Dense
from tensorflow.keras.layers import LSTM, GRU, SimpleRNN, TimeDistributed, Bidirectional, ConvLSTM2D, Dropout
from tensorflow.keras.models import model_from_json
#定义参数
model_number = str()
learning_rate = 0.01
dropout_rate = 0
layer_size = 1
units = 1
activation_function = 'relu'
loss_function = 'mae'
epoch = 1
batch_size = 1
models = []
optimizers = ''
#优化器选择
if optimizers == 'SGD':
optimizer = tf.keras.optimizers.SGD(learning_rate=learning_rate, clipnorm=1)
elif optimizers == 'Adam':
optimizer = tf.keras.optimizers.Adam(learning_rate=learning_rate, clipnorm=1)
elif optimizers == 'Adamax':
optimizer = tf.keras.optimizers.Adamax(learning_rate=learning_rate, clipnorm=1)
elif optimizers == 'Adagrad':
optimizer = tf.keras.optimizers.Adagrad(learning_rate=learning_rate, clipnorm=1)
elif optimizers == 'RMSprop':
optimizer = tf.keras.optimizers.RMSprop(learning_rate=learning_rate, clipnorm=1)
elif optimizers == 'Adadelta':
optimizer = tf.keras.optimizers.Adadelta(learning_rate=learning_rate, clipnorm=1)
elif optimizers == 'Nadam':
optimizer = tf.keras.optimizers.Nadam(learning_rate=learning_rate, clipnorm=1)
elif optimizers == 'Ftrl':
optimizer = tf.keras.optimizers.Ftrl(learning_rate=learning_rate, clipnorm=1)
return_sequence = Truedef fit_model(layer_size, X_train, X_test, y_train_df, batch_size, epoch,
units, activation_function, loss_function, optimizer, layer):
if layer_size == 1: # 1 gizli katman varsa
model = Sequential()
if layer[0] == 'GRU':
model.add(
GRU(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2])))
model.add(Dropout(dropout_rate))
elif layer[0] == 'LSTM':
model.add(
LSTM(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2])))
model.add(Dropout(dropout_rate))
elif layer[0] == 'SimpleRNN':
model.add(
SimpleRNN(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2])))
model.add(Dropout(dropout_rate))
elif layer_size == 2: # 2 gizli katman varsa
model = Sequential()
first_layer = layer[0][0]
second_layer = layer[0][1]
if (first_layer == 'GRU') and (second_layer == 'GRU'):
print("GRU GRU")
model.add(
GRU(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(GRU(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif (first_layer == 'LSTM') and (second_layer == 'LSTM'):
model.add(
LSTM(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(LSTM(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif (first_layer == 'SimpleRNN') and (second_layer == 'SimpleRNN'):
model.add(
SimpleRNN(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(SimpleRNN(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif (first_layer == 'GRU') and (second_layer == 'LSTM'):
model.add(
GRU(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(LSTM(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif (first_layer == 'GRU') and (second_layer == 'SimpleRNN'):
model.add(
GRU(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(SimpleRNN(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif (first_layer == 'LSTM') and (second_layer == 'SimpleRNN'):
model.add(
LSTM(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(SimpleRNN(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif (first_layer == 'LSTM') and (second_layer == 'GRU'):
model.add(
LSTM(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(GRU(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif (first_layer == 'SimpleRNN') and (second_layer == 'GRU'):
model.add(
SimpleRNN(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(GRU(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif (first_layer == 'SimpleRNN') and (second_layer == 'LSTM'):
model.add(
SimpleRNN(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(LSTM(units, activation=activation_function))
model.add(Dropout(dropout_rate))
else: # 4 gizli katman varsa
model = Sequential()
if layer[0] == 'GRU':
model.add(
GRU(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(GRU(units, activation=activation_function, return_sequences=True))
model.add(GRU(units, activation=activation_function, return_sequences=True))
model.add(GRU(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif layer[0] == 'LSTM':
model.add(
LSTM(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(LSTM(units, activation=activation_function, return_sequences=True))
model.add(LSTM(units, activation=activation_function, return_sequences=True))
model.add(LSTM(units, activation=activation_function))
model.add(Dropout(dropout_rate))
elif layer[0] == 'SimpleRNN':
model.add(
SimpleRNN(units,
activation=activation_function,
input_shape=(X_train.shape[1], X_train.shape[2]),
return_sequences=True))
model.add(Dropout(dropout_rate))
model.add(SimpleRNN(units, activation=activation_function, return_sequences=True))
model.add(SimpleRNN(units, activation=activation_function, return_sequences=True))
model.add(SimpleRNN(units, activation=activation_function))
model.add(Dropout(dropout_rate))
model.add(Dense(1))
model.compile(loss=loss_function, optimizer=optimizer)
history = model.fit(X_train,
y_train_df.to_numpy(),
epochs=epoch,
batch_size=batch_size,
validation_data=(X_test, y_test_df.to_numpy()),
verbose=1,
shuffle=False)
return history, model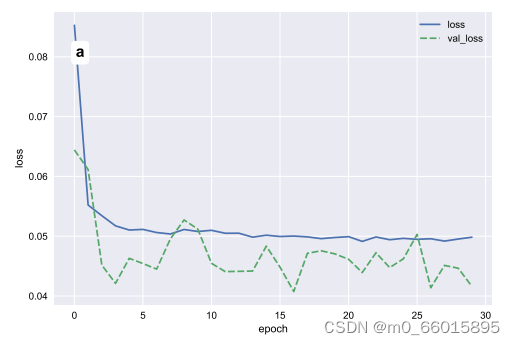

结论
综上所述,本研究通过整合遗传算法的HPO技术与深度学习模型,成功地提高了PM2.5浓度预测的准确性。实验结果表明,经过遗传算法优化后的模型相较于默认参数设置和随机搜索算法,在测试集和训练集上均展现出显著更优的表现。此外,该方法在处理实际存在的不完整、错误或异常数据方面也显示出了良好的鲁棒性,这对于现实世界应用尤为重要。未来,这种集成方法有望在其他环境监测和空气质量管理领域得到更广泛的应用。