还是来源于之前那篇cancer cell,小伙伴对于其中的一些分析方式比较感兴趣,之前没有提及,所以分享一下。原文结果是这样描述的”Notably, based on RNA-seq ontology graphic user environment (ROGUE)......our clusters demonstrated high internal homogeneity......",提到了一种方法ROGUE,可以判断细胞的同质性,或者说细胞群的纯度。
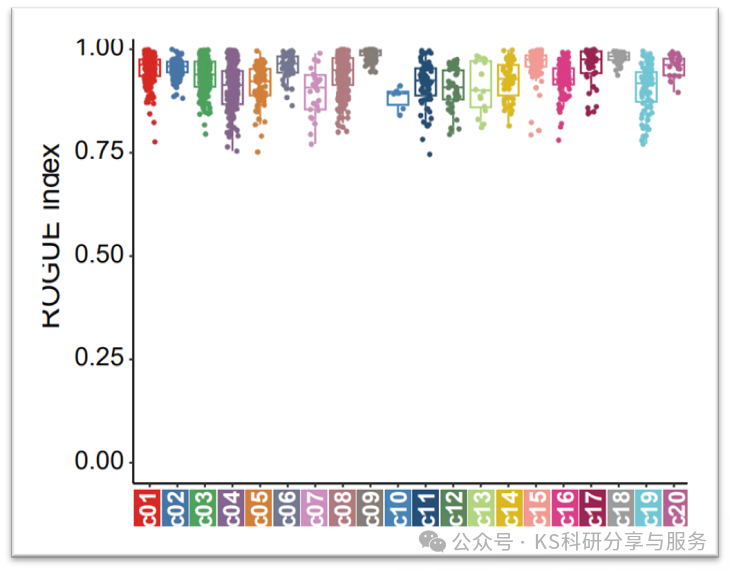
(reference:Cross-tissue human fibroblast atlas reveals myofibroblast subtypes with distinct roles in immune modulation)
看了一下,包出来很久了,2020年,是张泽民院士团队的,他们真的对单细胞领域贡献了好多方法。包的原文发表在Nature communications上,参考:An entropy-based metric for assessing the purityof single cell populations。这个包用于准确量化单细胞RNA测序(scRNA-seq)数据中鉴定的细胞簇的纯度。作者证明ROGUE广泛适用,能够对各种模拟和真实数据集中的簇纯度进行准确、敏感和稳健的评估。他们表明ROGUE可以识别额外的细胞亚型,并有助于检测特定亚群中的精确生物信号。感觉说了又好像没说:那具体有啥用呢?就不套用官方的话了,用我自己的理解说一下,不对的地方还请批评指正。主要有两个方面可应用:
其一:ROGUE值可以用于识别纯度较高的细胞亚型。这应该是大多数人会遇到的一个问题,提取大类细胞做亚群鉴定,到底分几群才合适呢?很多人可能是佛系的听天由命。而ROGUE值恰好可以为我们提供一个参考判断。ROGUE越高,越接近1,表明细胞群越纯,反之表示细胞群异质性比较高,这个群体还可再细分,这样我们可以分离得到一些亚群。
其二:ROGUE可以用于评估批次效应的影响。这个主要针对多数据集的整合,或者不同来源数据整合,比如我们见过一篇文献就是多个公共数据库数据整合,建设审稿人问你如何确定不同数据集批次效应有无去除,单纯的放一个UMAP图可能说服不了他,那么就可以搬出ROUGE算法,计算每种celltype在不同来源/不同sample中的纯度,如果ROUGE高,细胞群体纯度高,批次效应弱!
分析超级简单,步骤不多,首先安装包:
#github链接:https://kkgithub.com/PaulingLiu/ROGUE#教程链接:https://htmlpreview.github.io/?https://kkgithub.com/PaulingLiu/ROGUE/blob/master/vignettes/ROGUE_Tutorials.html#install.packages("tidyverse") 这个是依赖包,之前没有的话先安装if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")devtools::install_github("PaulingLiu/ROGUE")#下载安装不成功也可以本地安装,本地包下载地址https://codeload.github.com/PaulingLiu/ROGUE/legacy.tar.gz/HEAD
加载数据(我们演示的数据时一个Epi亚群初步分群的数据),提取矩阵和metadata:过滤低质量细胞和表达量低的基因。
setwd('D:\\KS项目\\公众号文章\\ROUGE单细胞纯度分析')#安装包# if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")# devtools::install_github("PaulingLiu/ROGUE")library(Seurat)library(ROGUE)library(tidyverse)library(ggplot2)library(ggrastr)#-------------------------------------------------------------------------------expr <- GetAssayData(Epi, assay = 'RNA',layer = 'counts') %>% as.matrix()meta <- Epi@meta.data#filter genes and cellsexpr <- matr.filter(expr, min.cells = 10, min.genes = 10)
计算表达熵模型,这是后续的基础:
ent.res <- SE_fun(expr)SEplot(ent.res)
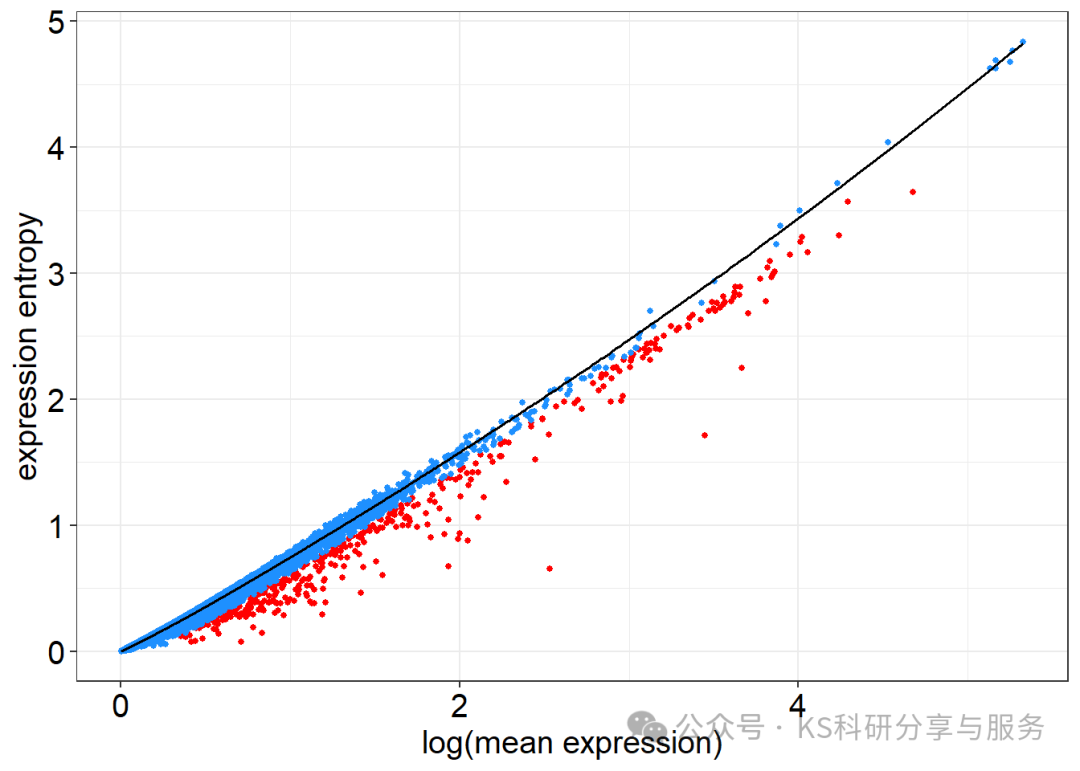
ROGUE calculation,这个是针对整个Epi细胞群体的计算,得到的最终值是0.3很低,说明Epi群体异质性很大,这个是符合的,且不说Epi可以分亚群,我们这个演示数据的Epi包含的还是正常人和肿瘤病人的Epi,那自然异质性更大了。
rogue.value <- CalculateRogue(ent.res, platform = "UMI")#[1] 0.339205
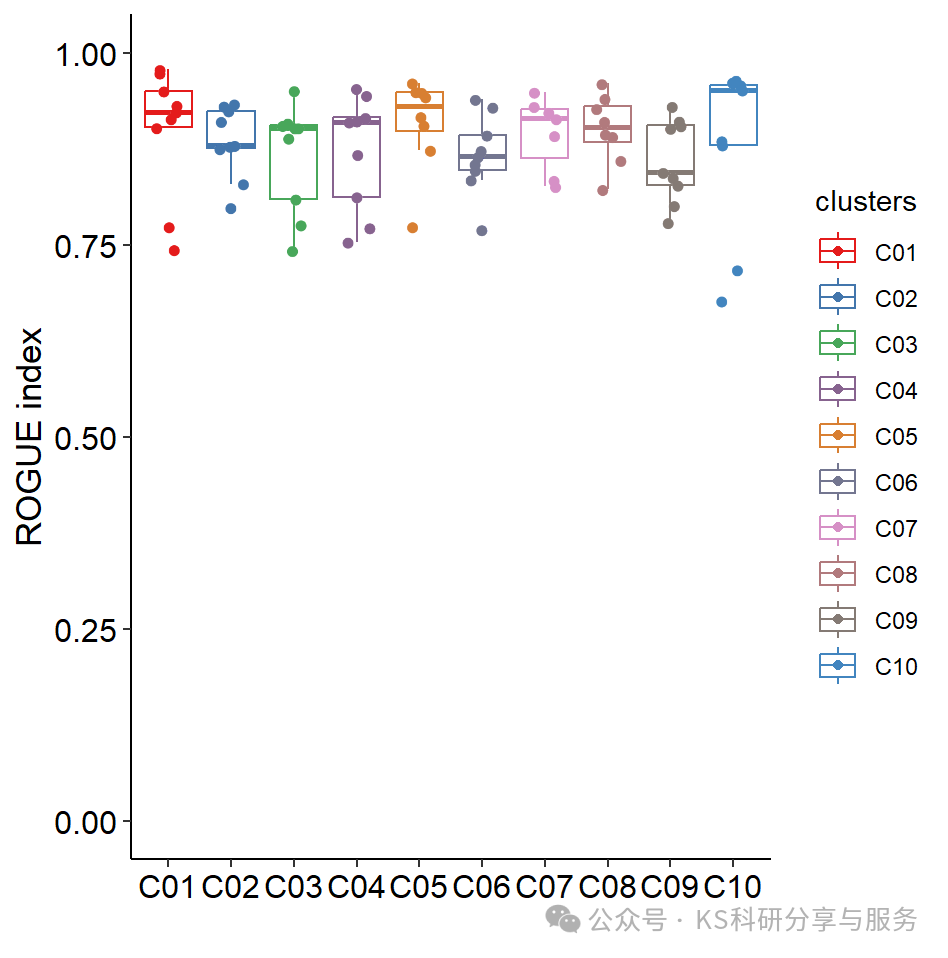
为了获得每个聚类的准确纯度估计值,计算不同样本中每种细胞类型的ROGUE值。并用箱线图可视化结果!
rogue.res <- rogue(expr, labels = meta$seurat_clusters, samples = meta$orig.ident, platform = "UMI", span = 0.6)
#这组颜色来源于cancer cell,可以收藏myColor <- c("#E41B1B", "#4376AC", "#48A75A", "#87638F", "#D87F32", "#737690", "#D690C6","#B17A7D", "#847A74", "#4285BF","#204B75", "#588257", "#B6DB7B", "#E3BC06", "#FA9B93", "#E9358B", "#A0094E", "#999999", "#6FCDDC", "#BD5E95")#宽数据转化为长数据格式,使用ggplot作图plotData <- rogue.res %>%tidyr::gather(key = clusters, value = ROGUE) %>%filter(!is.na(ROGUE))#散点箱线图ggplot(data = plotData, aes(clusters, ROGUE, color = clusters)) +geom_boxplot(outlier.shape = NA) +#添加boxgeom_jitter_rast(shape = 16, position = position_jitter(0.2)) +#添加抖动散点scale_color_manual(values = myColor) +theme_classic() +theme(axis.text = element_text(size = 12, colour = "black"),axis.title = element_text(size = 13, colour = "black")) +labs(x = "", y = "ROGUE index") +ylim(0, 1)
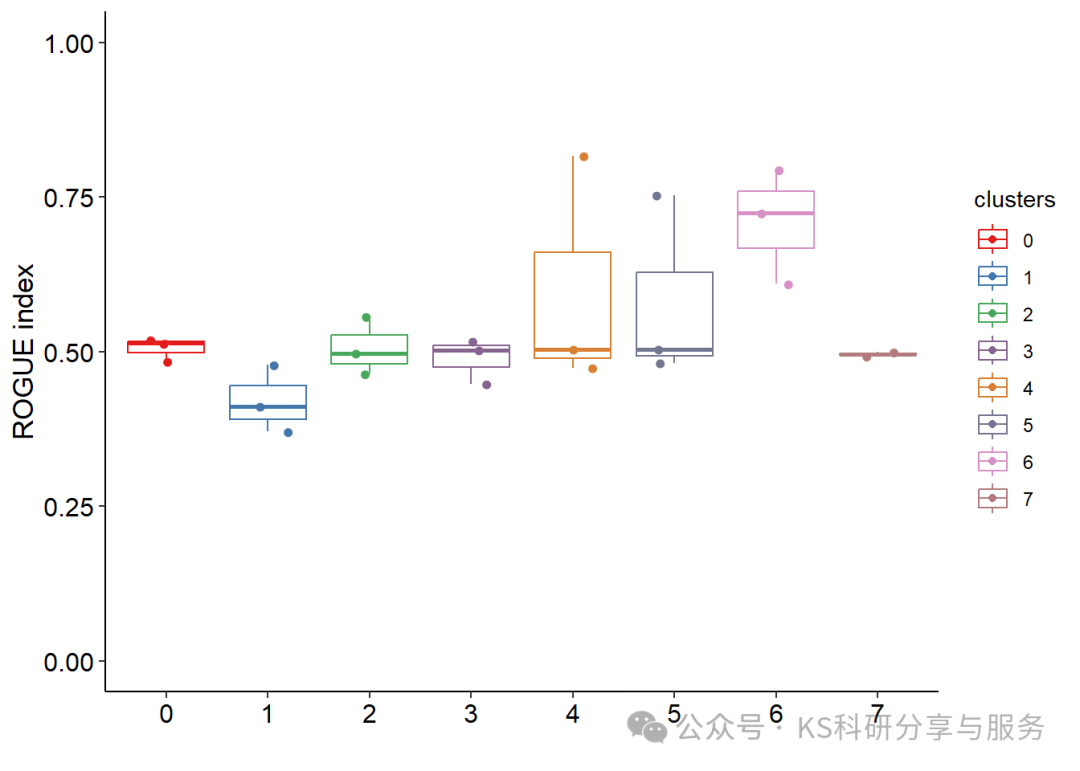
以上我们使用的是一个亚群分析的数据,可能有些例子举的不是很恰当,但是也可以作为参考。同时我们也演示另外一组数据,关于批次效应的数据演示这是一个大型数据集,合并了多个数据库不同来源的同一组织的单细胞数据,这里使用ROGUE验证一下。合并公共数据库最让人担心的问题不就是怕批次效应,或者数据集差异,最终得到错误结果。分析和前面一样,很简单。对于这个数据,最后我们计算了每个样本的cluster纯度,以及每个数据库下cluster纯度,发现ROGUE还可以,说明批次效应较小。
library(Seurat)library(ROGUE)library(tidyverse)library(ggplot2)library(ggrastr)# Rce <- subset(sce, sequencing=='scRNA_seq')#单细胞seurat obj#-------------------------------------------------------------------------------expr <- GetAssayData(Rce, assay = 'RNA',layer = 'counts') %>% as.matrix()meta <- Rce@meta.data#filter genes and cellsexpr <- matr.filter(expr, min.cells = 15, min.genes = 15)#-------------------------------------------------------------------------------ent.res <- SE_fun(expr)SEplot(ent.res)#-------------------------------------------------------------------------------#ROGUE calculationrogue.value <- CalculateRogue(ent.res, platform = "UMI")rogue.res.sample <- rogue(expr, labels = meta$celltype, samples = meta$orig.ident, platform = "UMI", span = 0.6)rogue.res.database <- rogue(expr, labels = meta$celltype, samples = meta$database, platform = "UMI", span = 0.6)write.csv(rogue.res.sample, file = 'rogue.res.sample.csv')write.csv(rogue.res.database, file = 'rogue.res.database.csv')
可视化:
rogue_sample <- read.csv('rogue.res.sample.csv', header = T,row.names = 1)myColor <- c("#E41B1B", "#4376AC", "#48A75A", "#87638F", "#D87F32", "#737690", "#D690C6", "#B17A7D", "#847A74", "#4285BF","#204B75", "#588257", "#B6DB7B", "#E3BC06", "#FA9B93", "#E9358B", "#A0094E", "#999999", "#6FCDDC", "#BD5E95")plot_rogue_sample <- rogue_sample %>%tidyr::gather(key = clusters, value = ROGUE) %>%filter(!is.na(ROGUE))ggplot(data = plot_rogue_sample, aes(clusters, ROGUE, color = clusters)) +geom_boxplot(outlier.shape = NA) +geom_jitter_rast(shape = 16, position = position_jitter(0.2)) +scale_color_manual(values = myColor) +theme_classic() +theme(axis.text = element_text(size = 12, colour = "black"),axis.title = element_text(size = 13, colour = "black")) +labs(x = "", y = "ROGUE index") +ylim(0, 1)
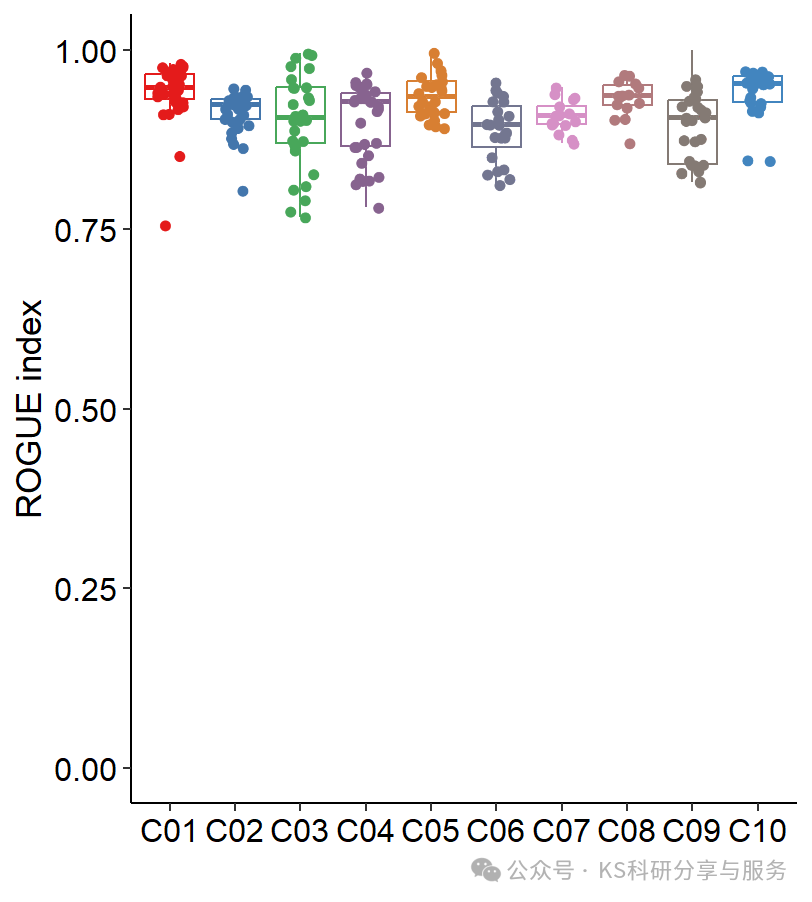
rogue_database <- read.csv('rogue.res.database.csv',header = T,row.names = 1)plot_rogue_database <- rogue_database %>%tidyr::gather(key = clusters, value = ROGUE) %>%filter(!is.na(ROGUE))ggplot(data = plot_rogue_database, aes(clusters, ROGUE, color = clusters)) +geom_boxplot(outlier.shape = NA) +geom_jitter_rast(shape = 16, position = position_jitter(0.2)) +scale_color_manual(values = myColor) +theme_classic() +theme(axis.text = element_text(size = 12, colour = "black"),axis.title = element_text(size = 13, colour = "black")) +labs(x = "", y = "ROGUE index") +ylim(0, 1)
觉得我们分享有些用的,点个赞再走呗!